Analysis of Antibody Aggregation on Cell Surfaces
Faculty MembersDr. Lydia TapiaPostdocsDr. Bruna JacobsonGraduate StudentsKasra ManaviJon David AlumniElijah JaffeAmanda Miner Jonah Spear Alan Kuntz Brittany Hoard CollaboratorsDr. Bridget S. WilsonRelated ProjectsProtein Binding Site Flexibility |
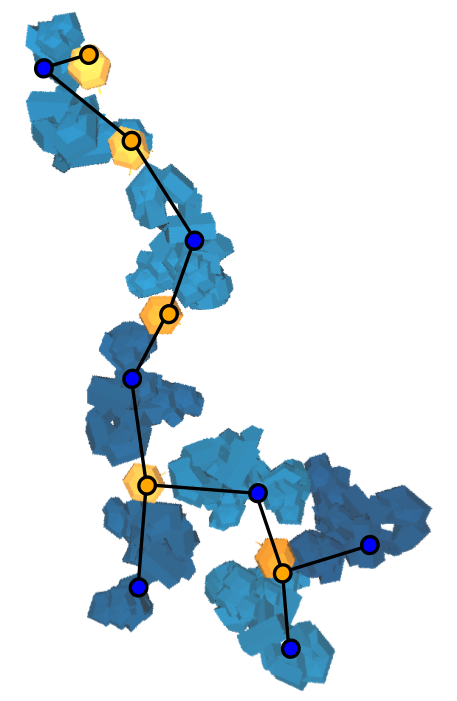
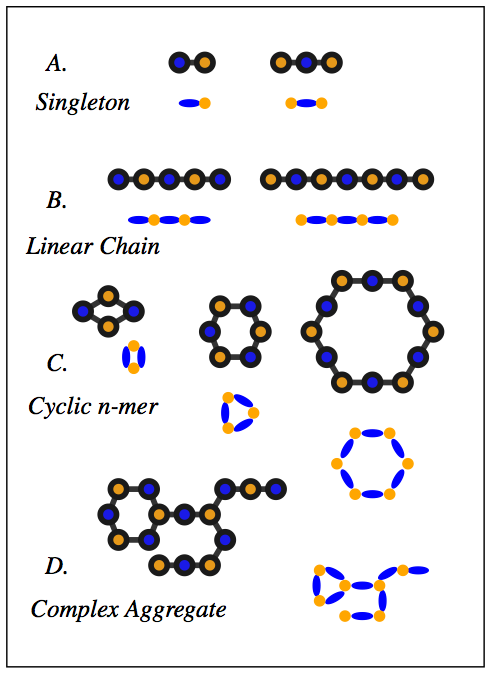
About 1,500 Americans die each year from anaphylatic shock caused by an immune response tiggered by antibody aggregation. IgE antibodies bound to cell-surface receptors, FceRI, crosslink through the binding of antigens on cell surfaces. This formation of aggregates is what stimulates mast cells and basophils in order to initiate an allergic response. Experimental studies have shown that the spatial organization of aggregated IgE-FceRI complexes affect transmembrane signaling that initiate these responses. 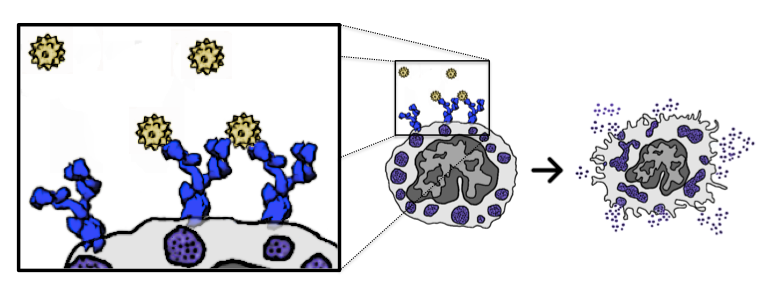 IgE antibodies (blue) crosslink through the binding of antigen (yellow) on the cell surface stimulates mast cells and basophils to release immune mediators (purple) We have developed methods to address modeling and analyzing this molecular data. To do this we created a Monte Carlo simulation to produce aggregate structures and created graph algorithms to classify these aggregates. To begin, we developed 3D models of a trivalent antigen and IgE-FceRI complex binding using relaxed constraints. These simplified models were generated from all-atom structures to reduce the complexity of the geometry. This reduces the computational complexity of the simulation to a rigid body problem which can be addressed using motion planning inspired techniques. 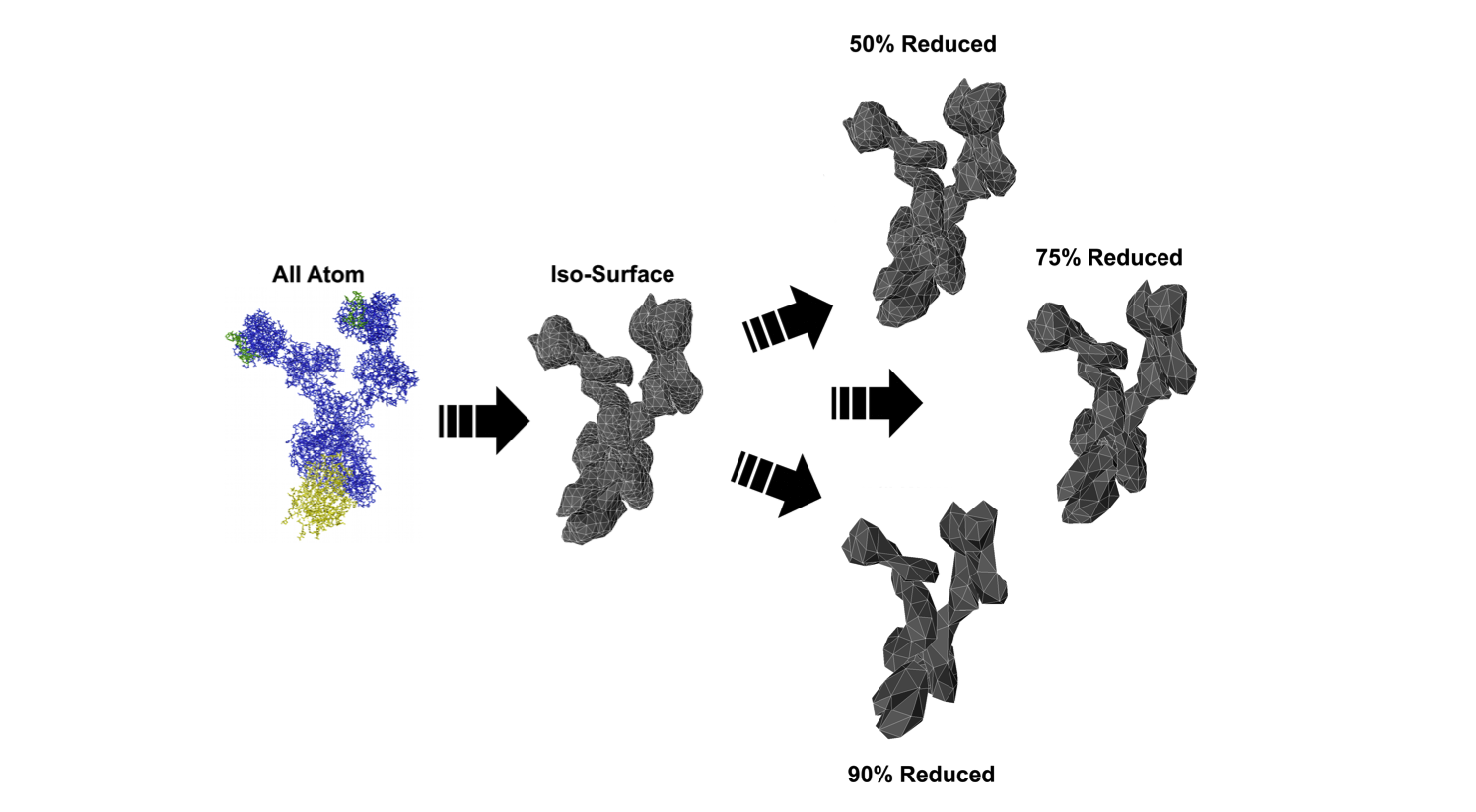 All atom molecular models (left) have their isosurfaces evaluated and used to generate a polygon model (middle)which can be reduced in complexity by simplifying the geometry (right) The models are simulated on a plane to capture movement of antibodies on the cell surface. The simulation uses kinetic rates and molecule speeds derived from experiments. The resulting aggregations are extracted from simulation runs.  Simulation of 90 Receptors and 180 Ligands In order to analyze the resulting structures, we introduce techniques to map molecular binding patterns to a graph structure. We came up with a classification scheme for aggregates using a depth-first search based algorithm:
This facilitates the analysis of aggregate structures because simple graph metrics, such as connected components and subgraph isomorphism, can be used to quickly quantify and analyze aggregate structure.
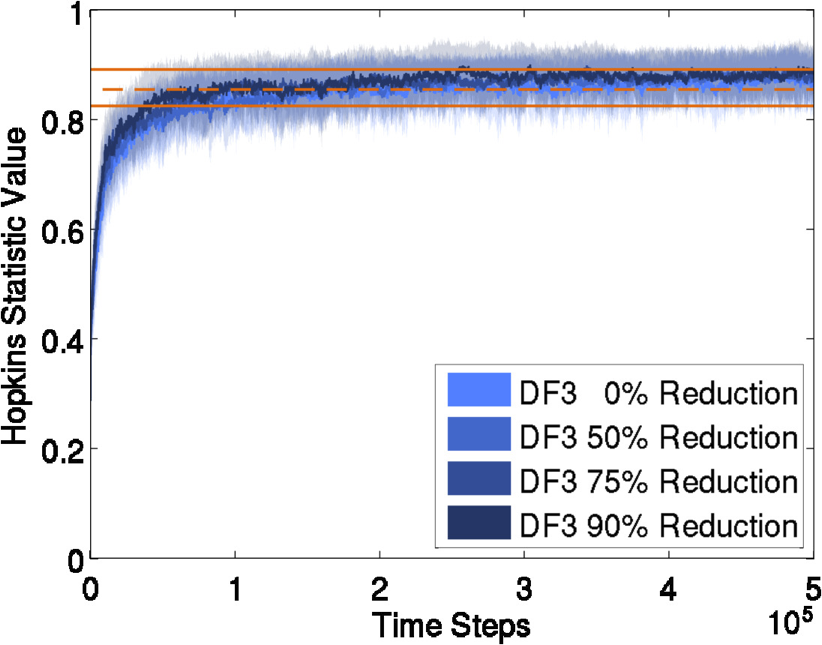
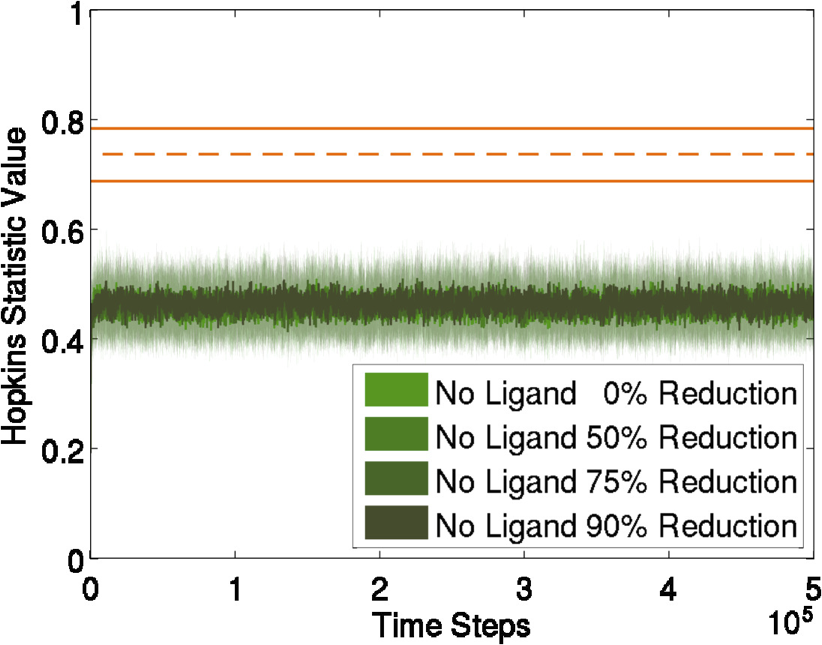
We study how model complexity affects molecular aggregation by analyzing its impact via two independent simulation techniques. First, we perform simulations using the techniques described above. Then, we approach this problem with a novel implementation of Rule-Based Modeling (RBM), a fast complementary simulation that does not explicitly represent geometry, and look for consistency in the results. We use binding site locations, surface curvature and steric hindrance induced by neighbor occupation to define our RBMs. 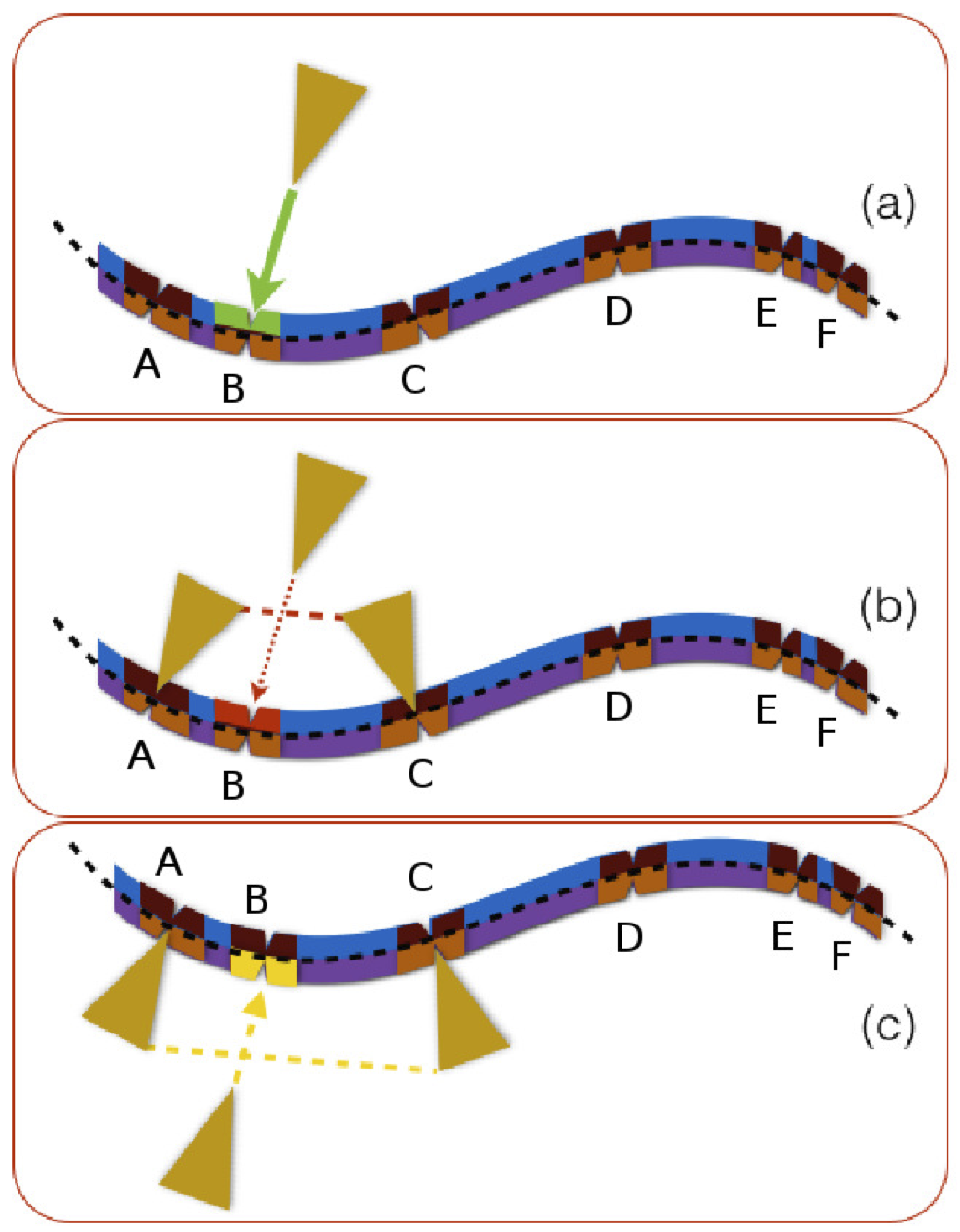 (a) No neighbors: receptors are free to bind, (b) Negative curvature reducing binding rate constant, and (c) Positive curvature with possible effect on binding We compare to experimental results when possible. For instance, a method used to evaluate aggregation is a measure of clustering (Hopkins statistic). We compare simulations with (blue) and without (green) ligand to experimentally derived values (orange).
We see similar values for the experiment with ligand. The difference seen in the experiment with no ligand is attributed to the nonhomogeneous nature of membranes. Molecular models of various resolutions are compared and little impact is seen from reduction. Publications & Papers(pdf, BibTex, abstract) (pdf, BibTex, abstract) (pdf, BibTex, abstract) |